Vacuum distillation is a method of distillation whereby the strain above the liquid mixture to be distilled is reduced to lower than its vapor strain (usually less than atmospheric strain) causing evaporation of essentially the most unstable liquid(s) (these with the bottom boiling factors).[1] This distillation methodology works on the principle that boiling happens when the vapor strain of a liquid exceeds the ambient stress. Vacuum distillation is used with or without heating the mixture.
 1 Laboratory-scale applications 1.1 Rotary evaporation
1 Laboratory-scale applications 1.1 Rotary evaporation
1.2 Distillation of excessive-boiling and/or air delicate supplies 1.2.1 Perkin triangle distillation set-up
1.2.2 Vacuum distillation set-up utilizing a brief-path head
2.1 Vacuum distillation in petroleum refining
Laboratory-scale purposes[edit]
Laboratory-scale vacuum distillation is used when liquids to be distilled have excessive atmospheric boiling points or chemically change at temperatures near their atmospheric boiling factors.[2][three][4] Temperature sensitive materials (akin to beta carotene) additionally require vacuum distillation to take away solvents from the mixture without damaging the product. Another reason vacuum distillation is used is that in comparison with steam distillation there is a decrease level of residue build up. That is vital in commercial applications where heat transfer is produced utilizing heat exchangers.
There are many laboratory applications for vacuum distillation as well as many varieties of distillation set-ups and apparatuses.
Safety is a vital consideration when using glassware as a part of the set-up. All of the glass elements must be carefully examined for scratches and cracks which could end in implosions when the vacuum is utilized. Wrapping as a lot of the glassware with tape as is practical helps to stop harmful scattering of glass shards in the occasion of an implosion.
Rotary evaporation[edit]
Rotary evaporation[5] is a type of vacuum distillation apparatus used to take away bulk solvents from the liquid being distilled. It is also utilized by environmental regulatory businesses for determining the quantity of solvents in paint, coatings and inks.[6]
Rotary evaporation set-ups embody an apparatus known as a Rotovap which rotates the distillation flask (typically called the still pot) to reinforce the distillation. Rotating the flask throws up liquid on the walls of the flask and thus increases the surface space for evaporation.
Heat is often utilized to the rotating distillation flask by partially immersing it in a heated bath of water or oil. Usually, the vacuum in such techniques is generated by a water aspirator or a vacuum pump of some sort.
Distillation of high-boiling and/or air sensitive materials[edit]
Some compounds have high boiling point temperatures in addition to being air sensitive. A simple laboratory vacuum distillation glassware set-up can be used, during which the vacuum can be replaced with an inert gas after the distillation is full.
Nonetheless, this is not a totally passable system if it is desired to collect fractions beneath a decreased pressure.
For better results or for very air sensitive compounds, both a Perkin triangle distillation set-up or a brief-path distillation set-up can be used.
Perkin triangle distillation set-up[edit]
The Perkin triangle set-up (Image 5) uses a sequence of Teflon valves to permit the distilled fractions to be isolated from the distillation flask with out the primary body of the distillation set-up being removed from both the vacuum or the heat source, and thus can stay in a state of reflux.
To do this, the distillate receiver vessel is first isolated from the vacuum by way of the Teflon valves.
The vacuum over the sample is then changed with an inert gas (corresponding to nitrogen or argon) and the distillate receiver can then be stoppered and removed from the system.
Vacuum distillation set-up utilizing a short-path head[edit]
Vacuum distillation of moderately air/water-sensitive liquid may be accomplished utilizing normal Schlenk-line techniques (Image 6). When assembling the set-up apparatus, the entire connecting strains are clamped so that they cannot pop off.
As soon as the apparatus is assembled, and the liquid to be distilled is within the nonetheless pot, the desired vacuum is established in the system by utilizing the vacuum connection on the brief-path distillation head. Care is taken to prevent potential “bumping” as the liquid within the nonetheless pot degases.
Whereas establishing the vacuum, the stream of coolant is started through the quick-path distillation head. Once the specified vacuum is established, heat is applied to the nonetheless pot.
If needed, the first portion of distillate might be discarded by purging with inert gas and altering out the distillate receiver.
When the distillation is complete: the heat is eliminated, the vacuum connection is closed, and inert gas is purged through the distillation head and the distillate receiver. Whereas beneath the inert gas purge, remove the distillate receiver and cap it with an air-tight cap. The distillate receiver might be saved under vacuum or beneath inert gas by utilizing the facet-arm on the distillation flask.
Industrial-scale purposes[edit]
Industrial-scale vacuum distillation[eight] has a number of advantages. Shut boiling mixtures could require many equilibrium phases to separate the important thing parts. One instrument to reduce the number of stages wanted is to make the most of vacuum distillation.[9] Vacuum distillation columns (as depicted in Figures 2 and three) usually used in oil refineries have diameters ranging as much as about 14 meters (46 toes), heights ranging as much as about 50 meters (164 toes), and feed charges ranging as much as about 25,four hundred cubic meters per day (160,000 barrels per day).
Vacuum distillation will increase the relative volatility of the key parts in many functions. The higher the relative volatility, the more separable are the 2 parts; this connotes fewer levels in a distillation column to be able to effect the same separation between the overhead and bottoms products. Lower pressures enhance relative volatilities in most programs.
A second benefit of vacuum distillation is the diminished temperature requirement at decrease pressures. For a lot of techniques, the products degrade or polymerize at elevated temperatures.
Vacuum distillation can enhance a separation by:
– Prevention of product degradation or polymer formation because of reduced stress resulting in decrease tower bottoms temperatures,
– Reduction of product degradation or polymer formation because of lowered imply residence time particularly in columns utilizing packing somewhat than trays.
– Increasing capability, yield, and purity.
Another benefit of vacuum distillation is the decreased capital value, on the expense of slightly extra working cost. Using vacuum distillation can scale back the height and diameter, and thus the capital price of a distillation column.
Vacuum distillation in petroleum refining[edit]
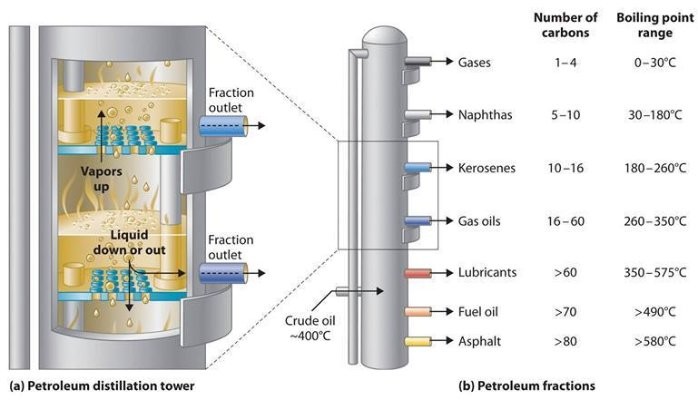
Petroleum crude oil is a complex mixture of hundreds of various hydrocarbon compounds generally having from 3 to 60 carbon atoms per molecule, though there could also be small quantities of hydrocarbons outdoors that vary.[10][11][12] The refining of crude oil begins with distilling the incoming crude oil in a so-called atmospheric distillation column operating at pressures slightly above atmospheric pressure.[8][10][11]
Vacuum distillation will also be referred as “low temperature distillation”
In distilling the crude oil, it is necessary to not subject the crude oil to temperatures above 370 to 380 °C as a result of the high molecular weight elements within the crude oil will bear thermal cracking and type petroleum coke at temperatures above that. Formation of coke would lead to plugging the tubes in the furnace that heats the feed stream to the crude oil distillation column. Plugging would additionally occur within the piping from the furnace to the distillation column in addition to within the column itself.
The constraint imposed by limiting the column inlet crude oil to a temperature of less than 370 to 380 °C yields a residual oil from the underside of the atmospheric distillation column consisting totally of hydrocarbons that boil above 370 to 380 °C.
To additional distill the residual oil from the atmospheric distillation column, the distillation must be performed at absolute pressures as little as 10 to 40 mmHg (also known as Torr) in order to restrict the operating temperature to less than 370 to 380 °C.
Figure 2 is a simplified course of diagram of a petroleum refinery vacuum distillation column that depicts the internals of the column and Figure 3 is a photograph of a large vacuum distillation column in a petroleum refinery.
The ten to 40 mmHg absolute pressure in a vacuum distillation column will increase the volume of vapor formed per volume of liquid distilled. The result’s that such columns have very massive diameters.[Thirteen]
Distillation columns such those in Photographs 1 and 2, might have diameters of 15 meters or extra, heights ranging up to about 50 meters, and feed rates ranging as much as about 25,four hundred cubic meters per day (160,000 barrels per day).
The vacuum distillation column internals should present good vapor-liquid contacting whereas, at the same time, maintaining a really low pressure enhance from the highest of the column prime to the bottom. Due to this fact, the vacuum column makes use of distillation trays only the place withdrawing merchandise from the aspect of the column (referred to as side draws). A lot of the column uses packing material for the vapor-liquid contacting because such packing has a decrease pressure drop than distillation trays. This packing material might be either structured sheet metallic or randomly dumped packing such as Raschig rings.
The absolute stress of 10 to 40 mmHg in the vacuum column is most frequently achieved by using multiple levels of steam jet ejectors.[14]
Many industries, other than the petroleum refining industry, use vacuum distillation on a much a smaller scale.
Molecular distillation[edit]
Molecular distillation is vacuum distillation below the strain of 0.01 torr[15] (1.Three Pa). 0.01 torr is one order of magnitude above excessive vacuum, where fluids are in the free molecular movement regime, i.e. the imply free path of molecules is comparable to the dimensions of the tools. The gaseous part no longer exerts important pressure on the substance to be evaporated, and consequently, rate of evaporation no longer will depend on stress. That’s, as a result of the continuum assumptions of fluid dynamics not apply, mass transport is governed by molecular dynamics moderately than fluid dynamics. Thus, a brief path between the recent surface and the chilly floor is critical, typically by suspending a hot plate covered with a film of feed next to a cold plate with a line of sight in between. Molecular distillation is used industrially for purification of oils.
Gallery[edit]
A easy quick path vacuum distillation apparatus
Kugelrohr – a short path vacuum distillation apparatus
Perkin triangle – for air-delicate vacuum distillation
Vacuum distillation apparatus
Steady distillation
Fractionating column
Fractional distillation
Kugelrohr
^ Laurence M. Harwood; Christopher J. Moody (13 June 1989). Experimental organic chemistry: Principles and Practice (Illustrated ed.). WileyBlackwell. pp. 147-149. ISBN 978-0-632-02017-1.
^ Distillation (CU Boulder Natural Chemistry Teaching Labs)
^ Vacuum Distillation: New Methodology for Analyzing Organic Chemicals in a wide array of Samples (United States Environmental Safety Agency)
^ What is vacuum distillation? (Argonne Nationwide Laboratory’s NEWTON Ask-A-Scientist)
^ Operation of a Rotary Evaporator (Rotovap) (from the website of the College of British Columbia)
^ SCAQMD Check technique 302-91
^ Energy Institute webpage page
^ a b Kister, Henry Z. (1992). Distillation Design (1st ed.). McGraw-Hill. ISBN zero-07-034909-6.
^ Karl Kolmetz, Andrew W. Sloley et al. (2004), Designing Distillation Columns for Vacuum Service, 11th India Oil and Gas Symposium and Worldwide Exhibition, September 2004, Mumbai, India (also revealed in Hydrocarbon Processing, Could 2005)
^ a b Gary, J.H. & Handwerk, G.E. Nigerian (1984). Petroleum Refining Technology and Economics (2nd ed.). Marcel Dekker, Inc. ISBN zero-8247-7150-eight.
^ a b Leffler, W.L. (1985). Petroleum refining for the nontechnical particular person (2nd ed.). PennWell Books. ISBN 0-87814-280-0.
^ James G, Speight (2006). The Chemistry and Know-how of Petroleum (Fourth ed.). CRC Press. 0-8493-9067-2.
^ Karl Kolmetz, Andrew W.
If you have any queries relating to exactly where and how to use Kinetic Energy, you can get hold of us at our own web-site.